We saw last day that the main repository for biomolecular structure (the PDB database) only has ~250,000 entries.
UnitProtKB (the main protein sequence database) has over 200 million entries!
AlphaFold
In this hands-on session we will utilize AlphaFold to predict protein structure from sequence (Jumper et al. 2021).
Without the aid of such approaches, it can take years of expensive laboratory work to determine the structure of just one protein. With AlphaFold we can now accurately compute a typical protein structure in as little as ten minutes.
This major breakthrough (Figure 1) promises to place Molecular Biology in a new era where we can visualize, analyze and interpret the structures and functions of all proteins.
The EBI AlphaFold Database
The EBI AlphaFold database contains lots of computed structure models. It is increasingly likely that the strucutre you are interested in is already in this database at < https://alphafold.ebi.ac.uk/ >
There are 3 major outputs from AlphaFold
A model of structure in PDB format
a pLDDT score: that tells us how confident the model is for a given residue in your protein (High values are good, above 70)
a PAE score that tells us about protein packing quality
If you can’t find a matching entry for the sequence you are interested in AFDB, you can run AlphaFold yourself…
Running AlphaFold
We will use ColabFold to run AlphaFold on our sequence < https://colab.research.google.com/github/sokrypton/ColabFold/ >
Figure from AlphaFold Here!
Interpreting Results
Custom analysis of resulting models
We can read all the AlphaFold results in to R and do more quantitative analysis than just viewing the structures in Mol-star:
Read all the PDB models:
library(bio3d)# File names for all PDB modelspdb_files <-list.files("hivpr_23119/", pattern =".pdb", full.names =TRUE)# Print our PDB file namesbasename(pdb_files)
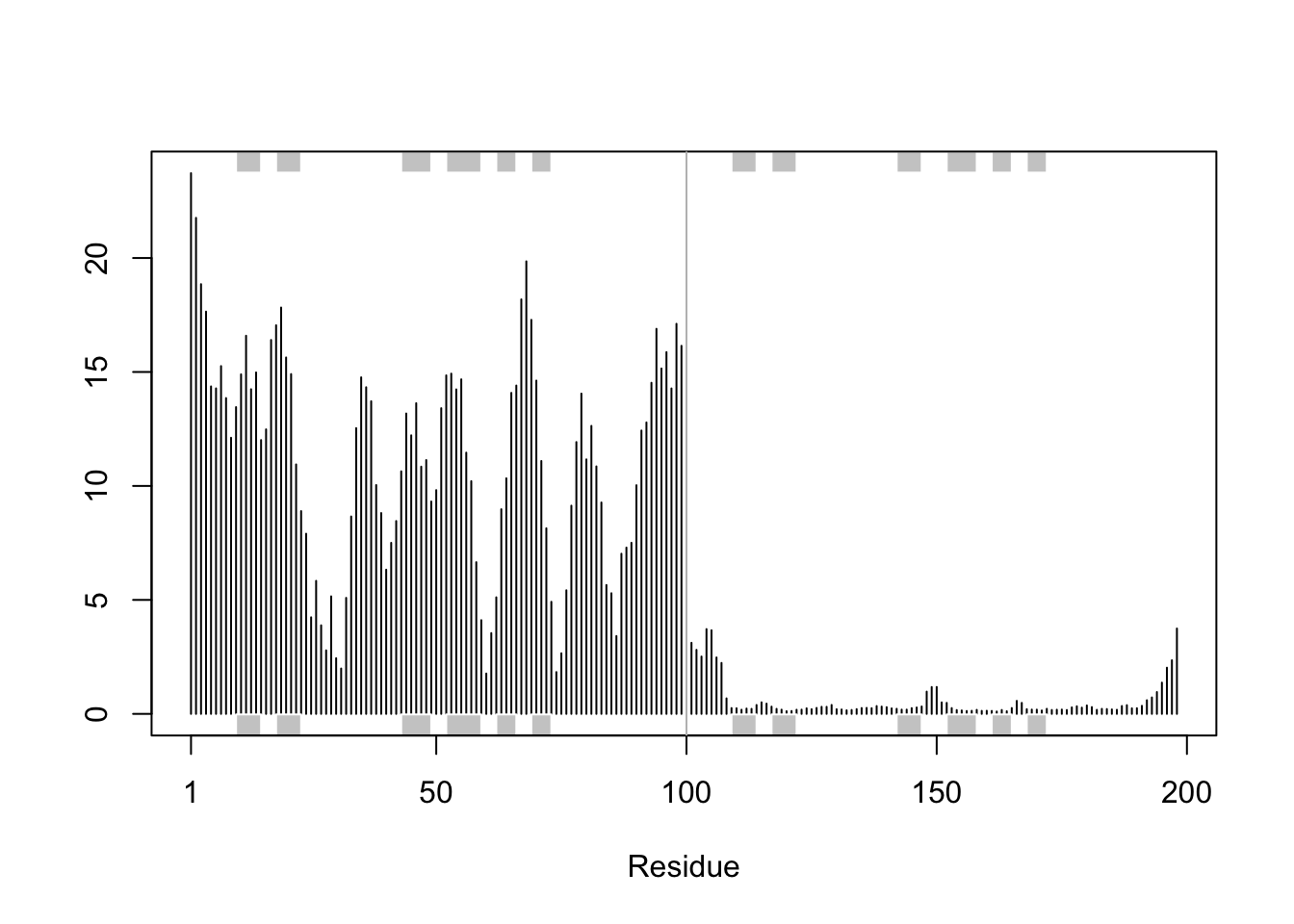
We can improve the superposition/fitting of our models by finding the most consistent “rigid core” common across all the models. For this we will use the core.find() function:
core <-core.find(pdbs)
core size 197 of 198 vol = 5310.035
core size 196 of 198 vol = 4635.834
core size 195 of 198 vol = 1812.524
core size 194 of 198 vol = 1109.016
core size 193 of 198 vol = 1035.296
core size 192 of 198 vol = 986.96
core size 191 of 198 vol = 941.365
core size 190 of 198 vol = 898.466
core size 189 of 198 vol = 857.922
core size 188 of 198 vol = 824.025
core size 187 of 198 vol = 793.081
core size 186 of 198 vol = 763.85
core size 185 of 198 vol = 740.429
core size 184 of 198 vol = 700.174
core size 183 of 198 vol = 673.821
core size 182 of 198 vol = 652.067
core size 181 of 198 vol = 634.066
core size 180 of 198 vol = 616.025
core size 179 of 198 vol = 599.771
core size 178 of 198 vol = 583.741
core size 177 of 198 vol = 568.863
core size 176 of 198 vol = 554.38
core size 175 of 198 vol = 536.073
core size 174 of 198 vol = 524.642
core size 173 of 198 vol = 510.527
core size 172 of 198 vol = 482.586
core size 171 of 198 vol = 464.833
core size 170 of 198 vol = 451.199
core size 169 of 198 vol = 435.724
core size 168 of 198 vol = 422.476
core size 167 of 198 vol = 412.705
core size 166 of 198 vol = 400.321
core size 165 of 198 vol = 389.367
core size 164 of 198 vol = 375.984
core size 163 of 198 vol = 364.58
core size 162 of 198 vol = 350.368
core size 161 of 198 vol = 338.444
core size 160 of 198 vol = 326.536
core size 159 of 198 vol = 315.655
core size 158 of 198 vol = 303.764
core size 157 of 198 vol = 292.94
core size 156 of 198 vol = 281.907
core size 155 of 198 vol = 272.908
core size 154 of 198 vol = 263.955
core size 153 of 198 vol = 254.522
core size 152 of 198 vol = 245.239
core size 151 of 198 vol = 231.669
core size 150 of 198 vol = 219.592
core size 149 of 198 vol = 211.974
core size 148 of 198 vol = 204.299
core size 147 of 198 vol = 194.165
core size 146 of 198 vol = 186.442
core size 145 of 198 vol = 178.703
core size 144 of 198 vol = 170.526
core size 143 of 198 vol = 163.019
core size 142 of 198 vol = 152.8
core size 141 of 198 vol = 143.077
core size 140 of 198 vol = 137.043
core size 139 of 198 vol = 131.686
core size 138 of 198 vol = 124.269
core size 137 of 198 vol = 117.206
core size 136 of 198 vol = 111.441
core size 135 of 198 vol = 104.8
core size 134 of 198 vol = 98.704
core size 133 of 198 vol = 94.662
core size 132 of 198 vol = 90.487
core size 131 of 198 vol = 87.458
core size 130 of 198 vol = 83.811
core size 129 of 198 vol = 79.337
core size 128 of 198 vol = 75.465
core size 127 of 198 vol = 71.717
core size 126 of 198 vol = 68.457
core size 125 of 198 vol = 64.79
core size 124 of 198 vol = 62.058
core size 123 of 198 vol = 58.244
core size 122 of 198 vol = 54.043
core size 121 of 198 vol = 49.43
core size 120 of 198 vol = 46.72
core size 119 of 198 vol = 43.468
core size 118 of 198 vol = 40
core size 117 of 198 vol = 37.229
core size 116 of 198 vol = 34.353
core size 115 of 198 vol = 31.473
core size 114 of 198 vol = 28.779
core size 113 of 198 vol = 26.188
core size 112 of 198 vol = 24.174
core size 111 of 198 vol = 22.314
core size 110 of 198 vol = 20.553
core size 109 of 198 vol = 18.935
core size 108 of 198 vol = 17.265
core size 107 of 198 vol = 15.517
core size 106 of 198 vol = 13.676
core size 105 of 198 vol = 12.402
core size 104 of 198 vol = 11.182
core size 103 of 198 vol = 10.242
core size 102 of 198 vol = 8.928
core size 101 of 198 vol = 8.18
core size 100 of 198 vol = 7.219
core size 99 of 198 vol = 6.1
core size 98 of 198 vol = 5.264
core size 97 of 198 vol = 4.359
core size 96 of 198 vol = 3.772
core size 95 of 198 vol = 3.183
core size 94 of 198 vol = 2.696
core size 93 of 198 vol = 2.414
core size 92 of 198 vol = 1.876
core size 91 of 198 vol = 1.611
core size 90 of 198 vol = 1.272
core size 89 of 198 vol = 1.038
core size 88 of 198 vol = 0.857
core size 87 of 198 vol = 0.686
core size 86 of 198 vol = 0.54
core size 85 of 198 vol = 0.44
FINISHED: Min vol ( 0.5 ) reached
# We can now use the identified core atom positions as a basis for a more suitable superposition and write out the fitted structures to a directory called corefit_structurescore.inds <-print(core, vol=0.5)
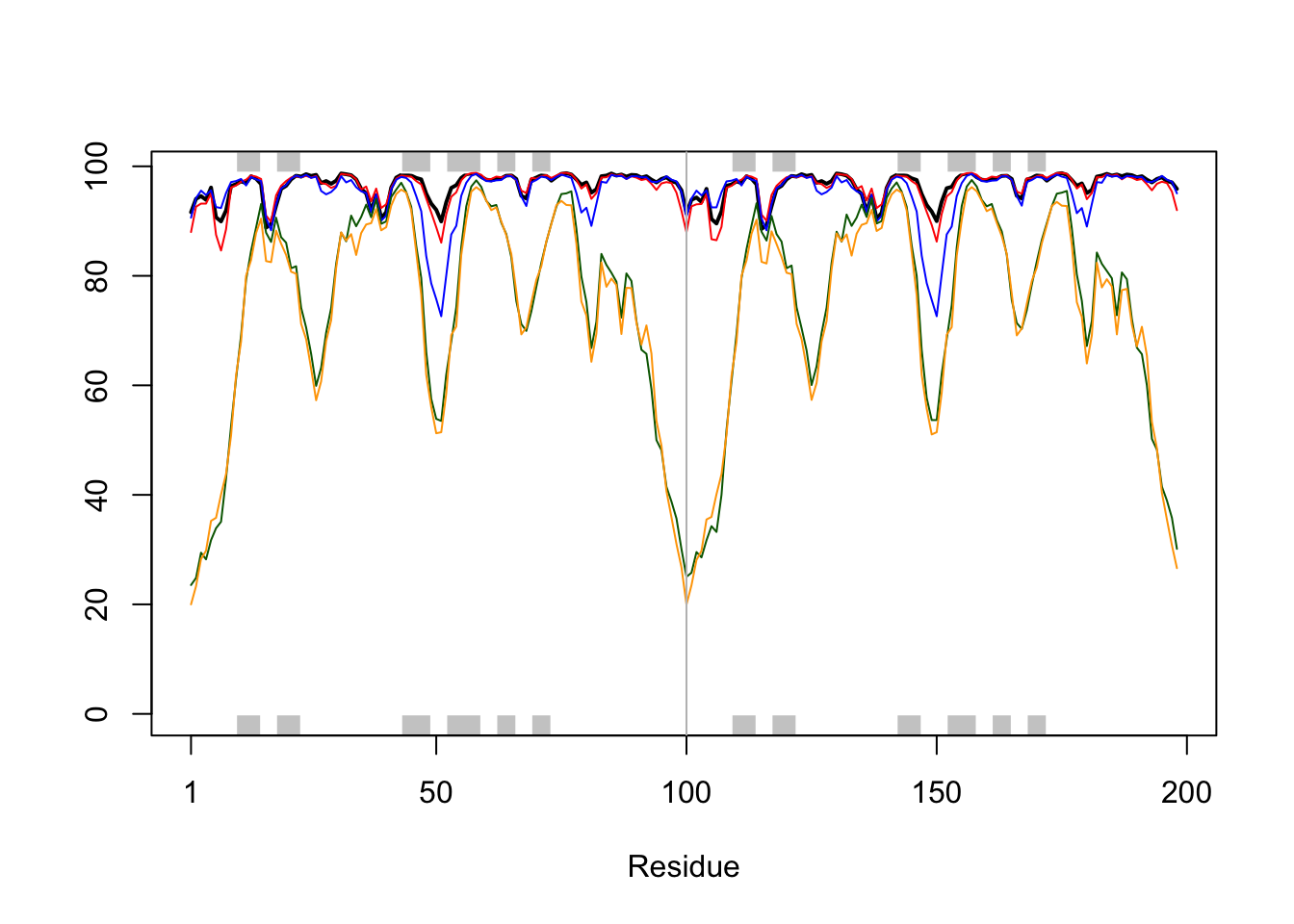
# Per-residue pLDDT scores # same as B-factor of PDB..head(pae1$plddt)
[1] 91.62 94.06 94.56 93.88 96.12 90.69
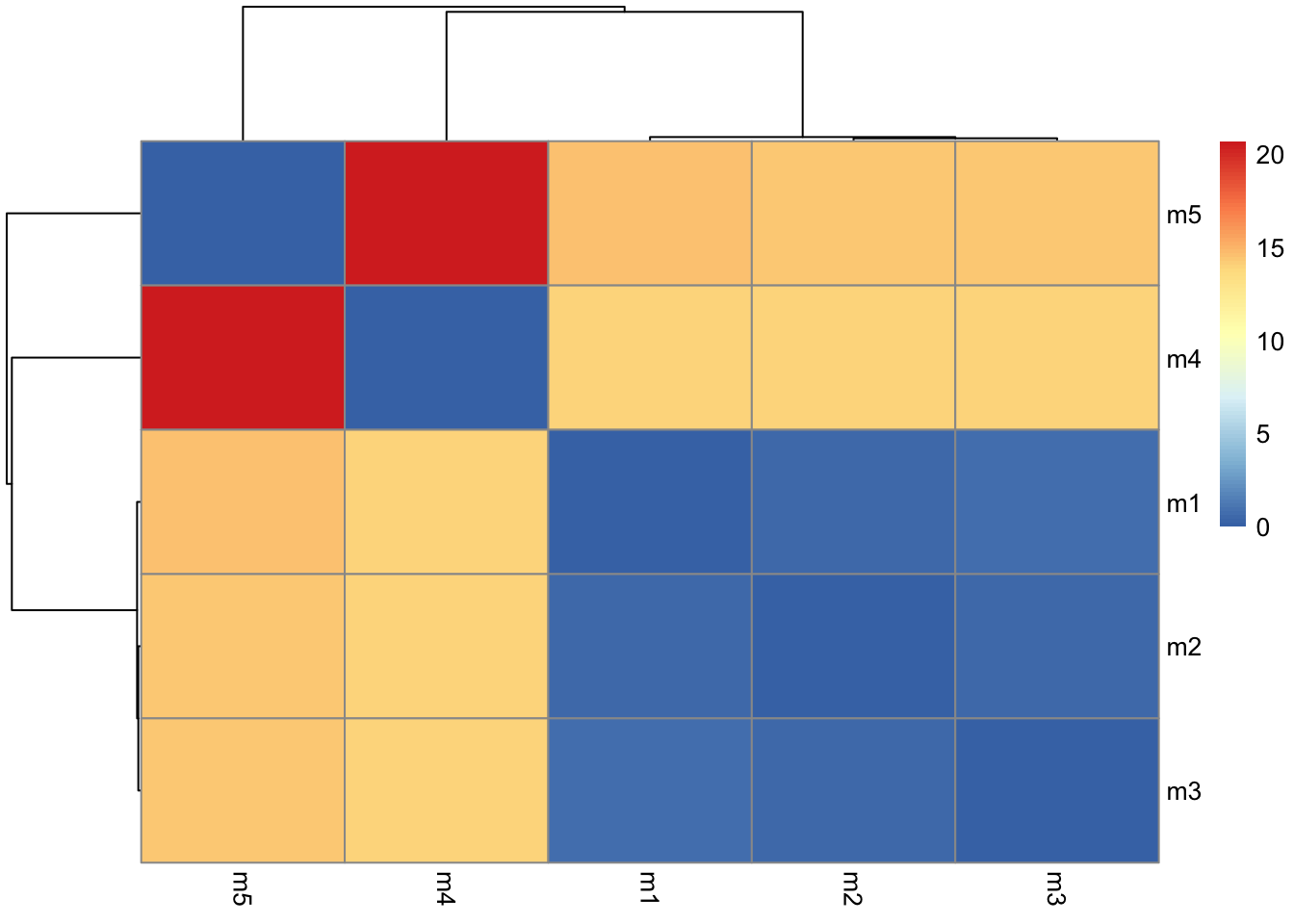
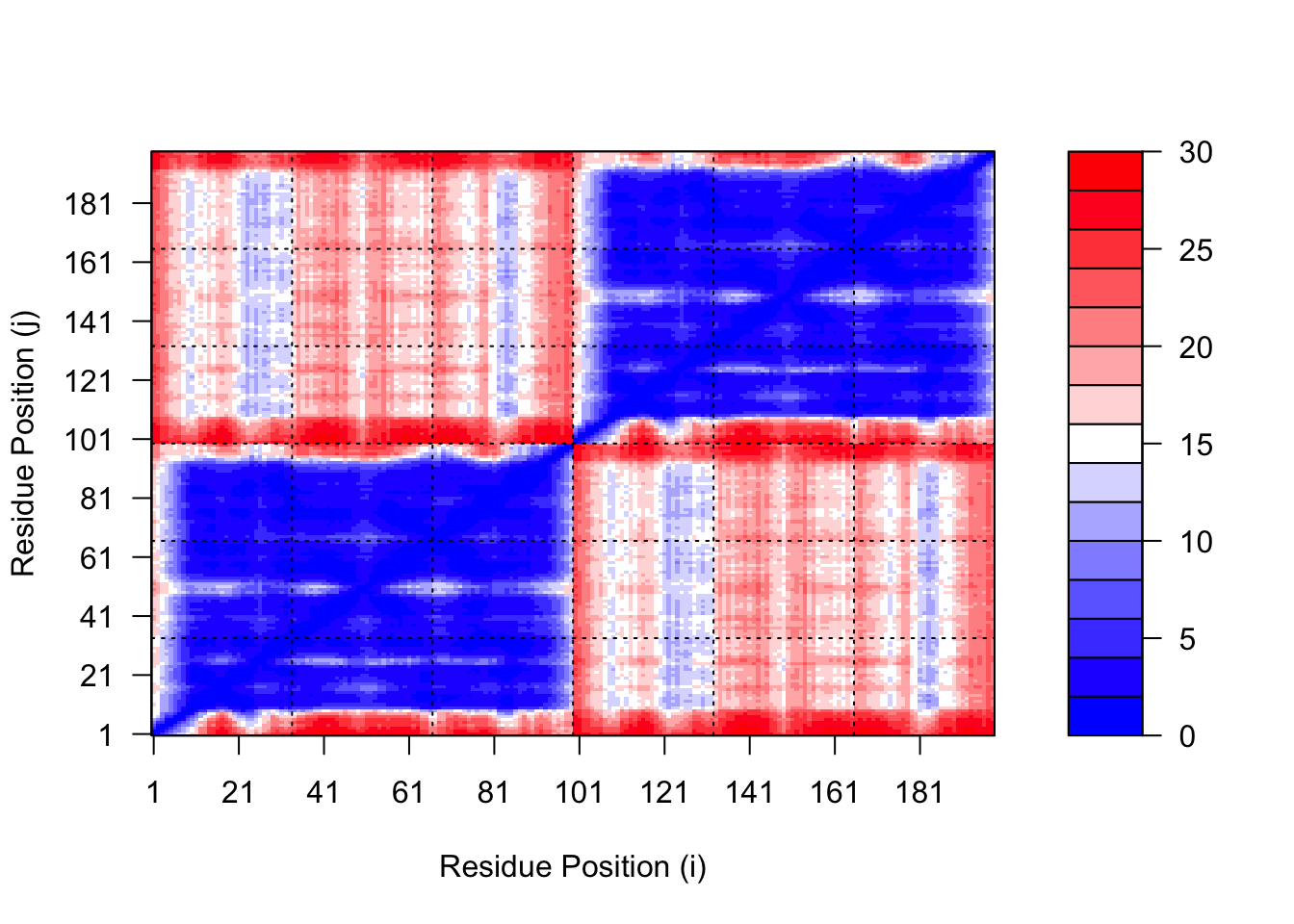
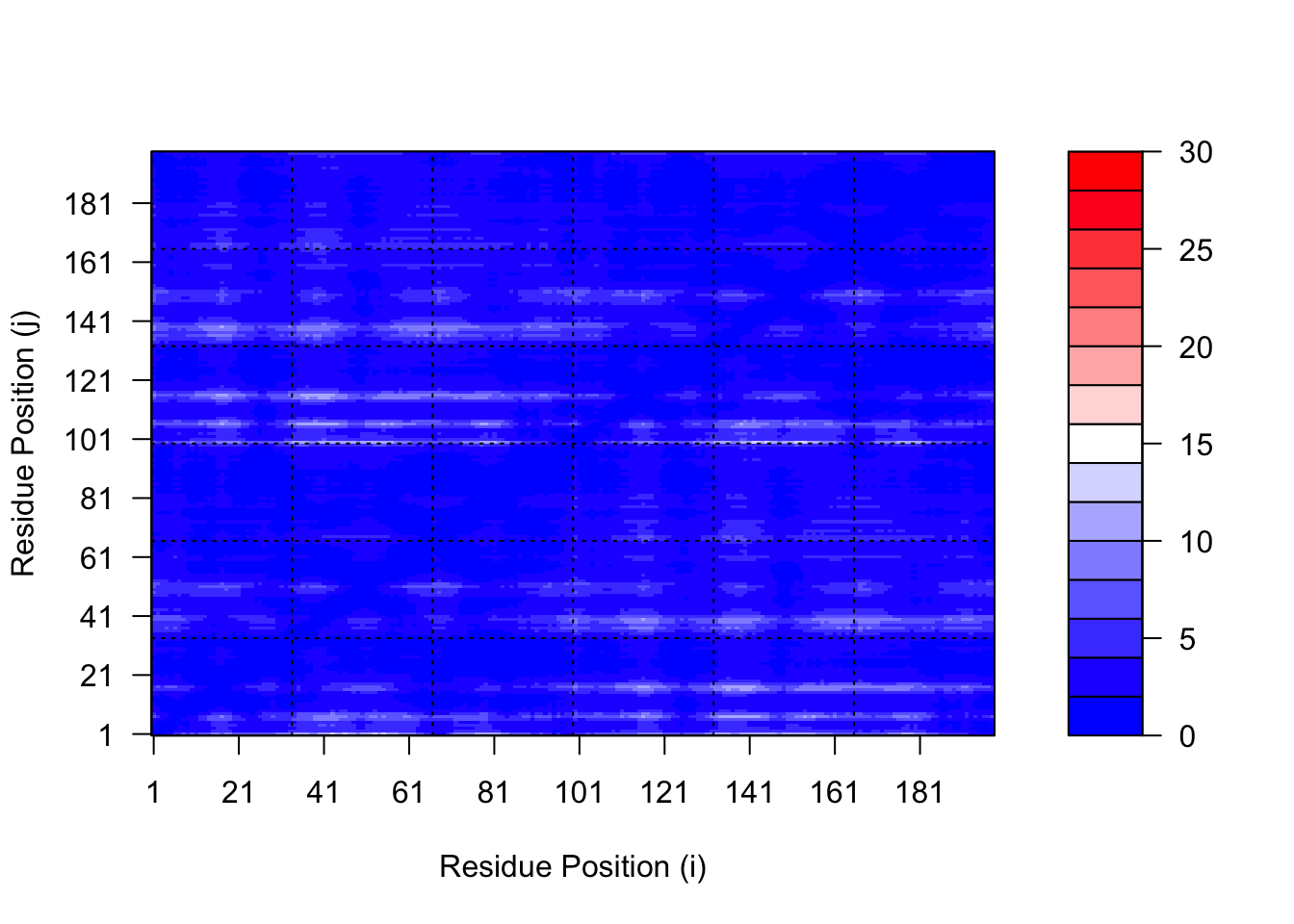
The maximum PAE values are useful for ranking models. Here we can see that model 5 is much worse than model 1. The lower the PAE score the better. How about the other models, what are thir max PAE scores?
pae1$max_pae
[1] 12.33594
pae5$max_pae
[1] 29.45312
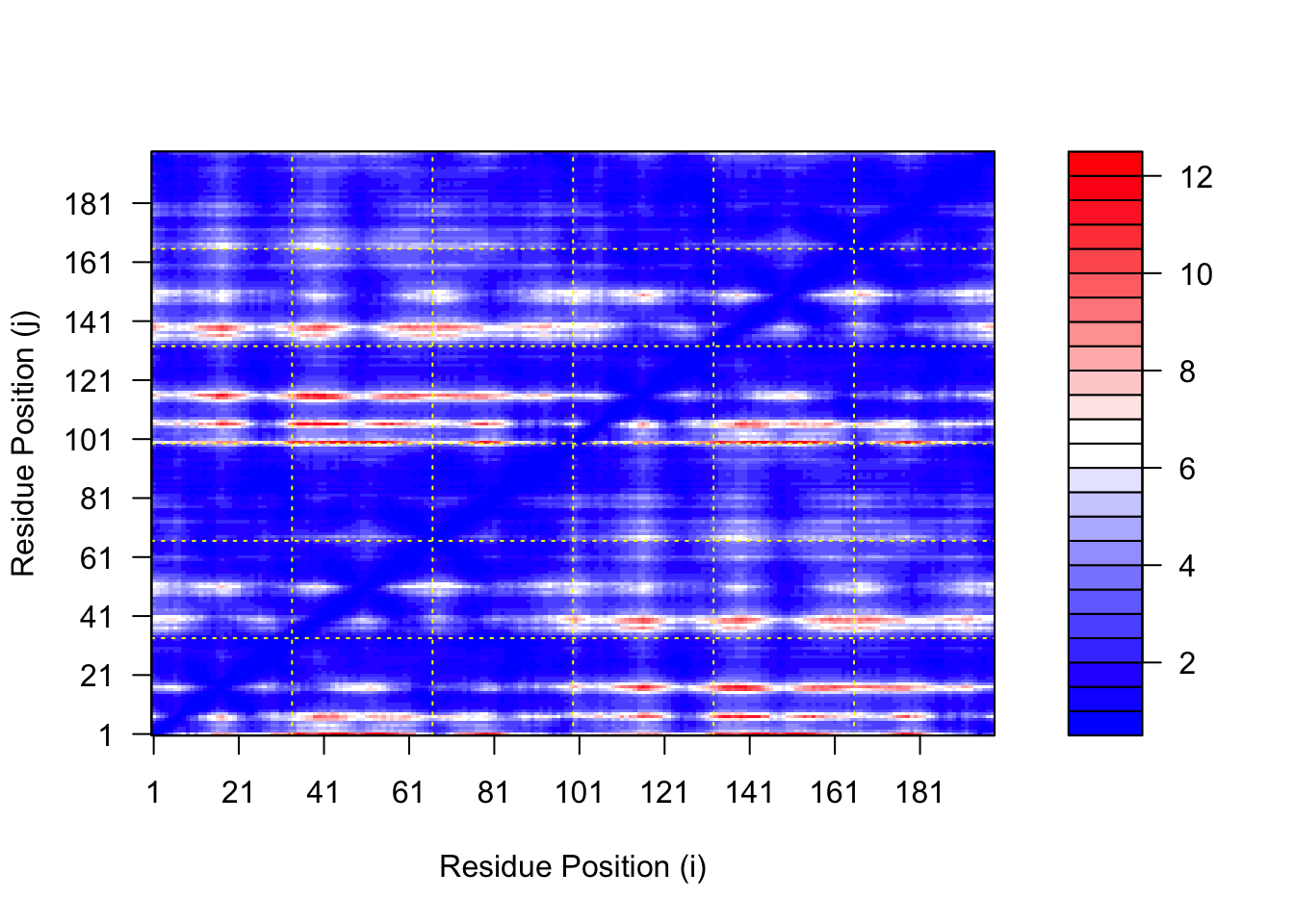
We can plot the N by N (where N is the number of residues) PAE scores with ggplot or with functions from the Bio3D package:
plot.dmat(pae1$pae, xlab="Residue Position (i)",ylab="Residue Position (j)")
plot.dmat(pae5$pae, xlab="Residue Position (i)",ylab="Residue Position (j)",grid.col ="black",zlim=c(0,30))
We should really plot all of these using the same z range. Here is the model 1 plot again but this time using the same data range as the plot for model 5:
plot.dmat(pae1$pae, xlab="Residue Position (i)",ylab="Residue Position (j)",grid.col ="black",zlim=c(0,30))